
Les facteurs de coagulation : quels rôles jouent-ils dans l’hémophilie ?
Les facteurs de coagulation sont des protéines du sang indispensables en cas de lésion vasculaire : les plaquettes et le facteur de Von Willebrand permettent de colmater la brèche et mettent fin au saignement.
Les facteurs VIII et IX sont particulièrement connus pour les maladies qu’ils provoquent en cas de déficit : respectivement l’hémophilie A et B.
l’hémostase primaire, qui correspond à la formation du clou plaquettaire venant obstruer la brèche vasculaire afin de bloquer le saignement en urgence ; suivie par l’hémostase secondaire, qui correspond à la formation d’un caillot sanguin plus solide faisant intervenir un processus complexe, la « cascade de coagulation ».
L’hémostase primaire ou formation du clou plaquettaire
Une réaction immédiate de l’organisme
Le rôle du facteur de Von Willebrand
L’hémostase secondaire ou formation du caillot sanguin
Le rôle des facteurs de coagulation
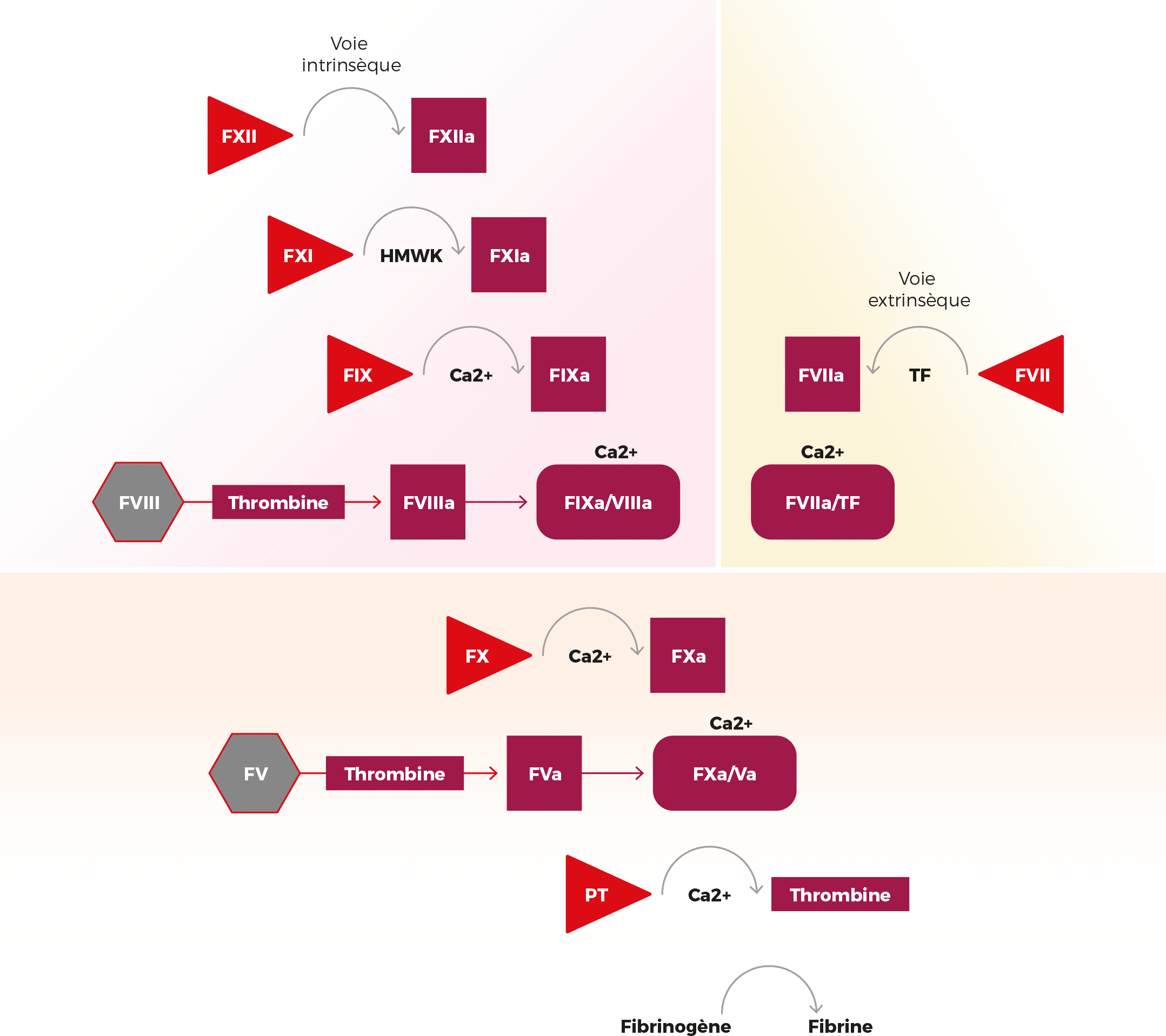
L’hémostase secondaire fait appel aux facteurs de coagulation. Il en existe 10, numérotés en chiffres romains (FI, FII, FV, FVII, FVIII, FIX, FX, FXI, FXII, FXIII). Ils circulent librement dans le sang sous une forme inactive et interviennent dès lors qu’une hémostase primaire a été initiée par les plaquettes sanguines pour faire face à une lésion vasculaire et prévenir un saignement. Ils doivent pour cela être activés par des enzymes, qui requièrent la présence de phospholipides et de calcium.
L’hémophilie, une pathologie de l’hémostase secondaire
La cascade de coagulation
Le clou plaquettaire formé lors de l’hémostase primaire est fragile et doit être renforcé rapidement : c’est le rôle des protéines de coagulation (2).
Elles vont enclencher une série de réactions biologiques interdépendantes conduisant à la production de fibrine venant se coller à la surface du clou plaquettaire pour le rendre imperméable et former le caillot sanguin.
L’initiation de la cascade de coagulation débute par la liaison du facteur VII sur le facteur tissulaire libéré lors de lésion du vaisseau.
Cela active le facteur VII qui, à son tour, va activer les facteurs X et IX. Une fois actif, le facteur Xa (a pour actif) va se lier au facteur Va présent à la surface des plaquettes puis entraîner la formation de thrombine.
Cette petite quantité de thrombine va activer les facteurs V, VIII et XI, qui vont ensuite pouvoir activer la cascade de coagulation et générer une plus grande quantité de thrombine et amplifier le procédé de coagulation.
Elément indispensable de l’hémostase primaire, le facteur de Von Willebrand intervient également lors de l’hémostase secondaire, pour protéger et prolonger la durée de vie d’un autre facteur de coagulation essentiel, le facteur VIII.

La maladie de Willebrand
Cette mutation se traduit soit par la fabrication en moindre quantité de la protéine, soit par la fabrication d’une forme altérée. Dans les deux cas, l’hémostase primaire initiant le processus de coagulation et l’hémostase secondaire sont inefficaces, ce qui se traduit par des saignements prolongés.
Il s’agit de la plus fréquente des maladies hémorragiques, atteignant jusqu’à 1 % de la population mondiale ; un nombre certainement largement sous-estimé en raison du caractère bénin des symptômes dans les formes modérées de la maladie (4).
Contrairement à l’hémophilie A et à l’hémophilie B, elle touche autant les filles que les garçons puisque le gène en cause est situé sur le chromosome 12 dont chaque enfant hérite de ses deux parents.


